The Xie lab started at Tsinghua University in Beijing since 2013. Our research centers on two key and fundamental questions – how does a totipotent embryo arise from two terminally differentiated gametes? How is the epigenetic information transmitted and reprogrammed from parents to the next generation? One of the best systems to answer these questions is early development, when parental epigenetic information is being passed on to the progenies and when the first cell fate commitment events take place which subsequently determine all future body plans. However, epigenetic reprogramming, especially at the level of chromatin, remains poorly understood given the extremely scarce experimental materials. We are dedicated to meet these challenges by first developing a series of ultra-low-input epigenetic analysis technologies, which are then applied to solve biological questions in combination with genetic tools. Our specific questions are described below:
Ⅰ. How are the epigenomes established and regulated in gametes?
Using ultra-sensitive ChIP-seq and CUT&RUN, we revealed landscapes of histone modifications in oocyte and sperm in zebrafish (Zhang et al., Mol. Cell, 2018), mouse (Zhang et al., Nature, 2016; Zheng et al., Molecular Cell, 2016; Xu et al., Nature Genetics, 2019), and human (Xia et al., Science 2019). Interestingly, histone modifications can adopt non-canonical forms and functions in oocytes that are distinct from those in somatic cells (Nature, Zhang et al., 2016).
In addition, we also examined chromatin structure in 3D space. By successfully developing an ultrasensitive tool to study 3D chromatin organization (sis-HiC), we showed how chromatin is folded during mammalian gametogenesis (Wang et al., Mol. Cell, 2019; Du et al., Mol. Cell, 2019).
We also revealed that genomic imprints, the parental epigenetic memories that are essential for development, are established through SETD2-mediated histone modification-DNA methylation crosstalks in oocytes (Xu et al., Nature Genetics, 2019). These marks are rather stable afterwards before they are removed again in primordial germ cells, where we found and how this is executed by a simple isoform switch of an epigenetic regulator TET1 (Zhang et al., Molecular Cell, 2016).
Ⅱ. How are the epigenomes reprogrammed during parental-to-zygotic transition?
By doing so, we are among the first to provide a global view of chromatin accessible states in preimplantation embryos at the DNA level in human and mouse (Wu et al., Nature, 2016; Wu et al., Nature, 2018). Furthermore, we sought to determine whether histone modifications from each parent can be inherited to early embryos in mouse (Zhang et al., Nature, 2016; Zheng et al., Molecular Cell, 2016; Xu et al., Nature Genetics, 2019), zebrafish (Zhang et al., Mol. Cell, 2018) and human (Xia et al., Science 2019). Finally, we showed how chromatin folds in 3D spaces after fertilization (Du et al., Nature, 2017).
Ⅲ. How are the embryonic epigenomes established and regulated?
Given epigenetic modifications are extensively removed upon fertilization, one critical question is how the embryonic epigenome is established afterwards. As exploratory studies, we examined how DNA methylomes (Zhang et al., Nature Genetics, 2018) and histone modifications (Xiang et al., Nature Genetics, 2019) are established during about 10 early mouse lineage specification during peri- and postimplantation development (E3.5 to E7.5). In addition, we also investigate the reprogramming of chromatin molecular architecture during postimplantation development (Zhang et al., Nature Genetics, 2018; Xiang et al., Nature Genetics, 2019).
Ⅳ. How the epigenomes are regulated in embryonic stem cells?
Besides early development, we also use embryonic stem cells to decipher mechanistic insights on epigenetic regulation of transcription. For example, we showed how developmental genes manage to avoid DNA methylation regardless whether they are transcribed or not (Li et al., Genome Biology, 2018).
For details, please refer to our reviews in chromatin reprogramming in gametogenesis and early development (Xu et al., Trends in Cell Biology, 2018; Zheng et al., Nature Reviews in Molecular Cell Biology, 2019).
Some of our exciting findings are described in detail below.
1. We revealed how chromatin is reprogrammed at the DNA levels upon fertilization in mammals (Wu et al., Nature, 2016).
The state of chromatin dictates fundamental cellular processes including gene expression. However, the regulatory circuitry that controls chromatin reprogramming in early development remains poorly understood. Accessible chromatin can expose key DNA sequences to transcription regulators leading to gene activation. ATAC-seq is a highly sensitive approach to detect open chromatin using as few as single cells (Buenrostro et al., 2013). However, our initial experiment showed that ATAC-seq libraries derived from early embryos were strongly contaminated by highly abundant mitochondrial DNA (up to 99%). Therefore, we developed CARM (Cas9-Assisted Removal of Mitochondrial DNA) to further improve ATAC-seq by specifically depleting the contaminating DNA using CRISPR/Cas9. Using this method, we found that early development employs unique gene regulatory mechanisms. For example, large accessible chromatin domains were found at active repeats prior to major zygotic genome activation (ZGA), with some spanning up to 110kb, indicating that chromatin may be highly permissive. Accessible chromatin then becomes restricted to promoters and enhancers after ZGA from late 2-cell stage. Using accessible chromatin states, we inferred putative TFs that may regulate transcription circuitry that govern early development. Our work demonstrated how chromatin is reprogrammed at the DNA level in early mammalian development.
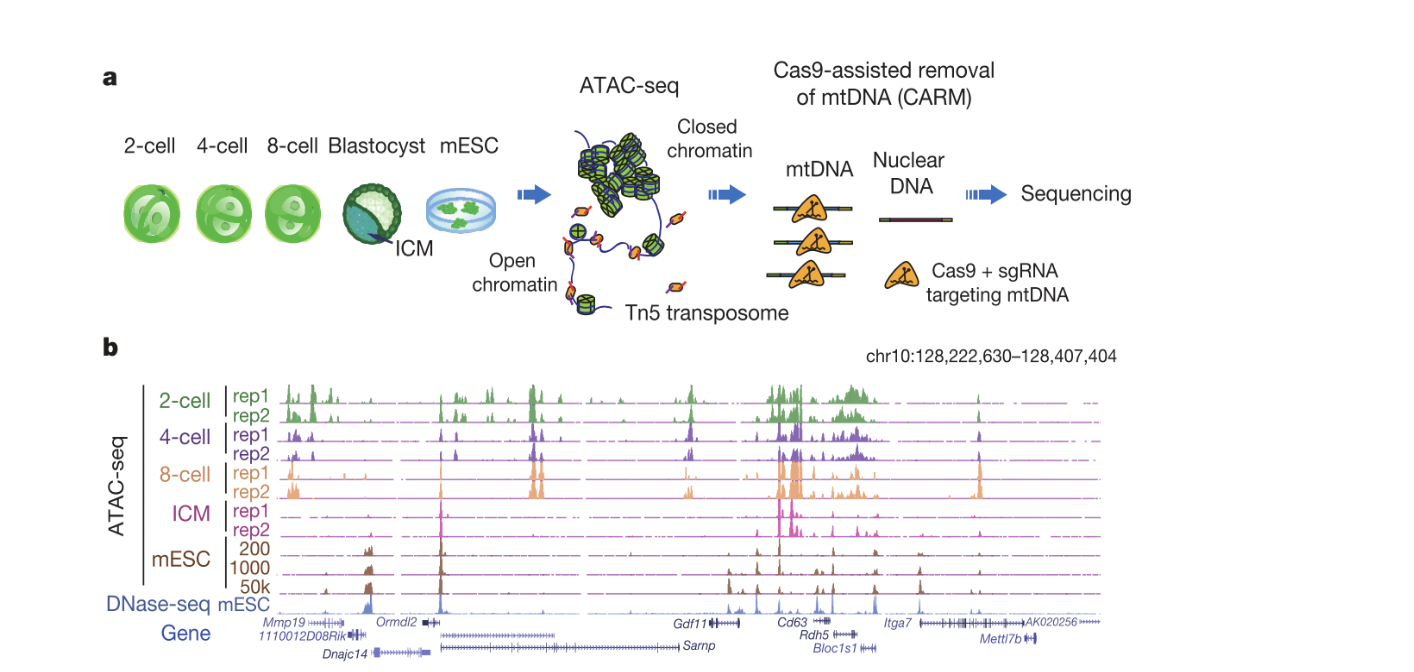
Figure 1. a, Schematic of ATAC-seq and CARM (Cas9-assisted removal of mtDNA) for probing accessible chromatin in mouse preimplantation embryos. b, The UCSC browser view shows enrichment of ATAC-seq in preimplantation embryos. Figures are adapted from Wu et al., Nature, 2016.
2. We demonstrated, for the first time, how histone modifications from each parent are transmitted to the next generation in mammals (Zhang et al., Nature, 2016).
Despite extensive investigations, whether histone modifications can be truly inherited to the next generation in mammals remains unknown. By developing an ultra-sensitive method STAR ChIP-seq, we revealed the landscapes of histone modifications in mouse early development. Interestingly, we found the histone mark H3K4me3 from the paternal genome is likely largely erased immediately after fertilization. Unexpectedly, in oocytes, H3K4me3 occurs primarily in broad domains in a large number of distal sites including intergenic regions. Such H3K4me3, which we termed as non-canonical H3K4me3 (ncH3K4me3), is then briefly inherited to early embryos prior to ZGA. Depleting H3K4me3 from mature oocytes results in defects in transcription silencing, raising a paradoxical possibility that H3K4me3 may be repressive in oocytes. Taken together, these results addressed a long-standing fundamental question in epigenetics – whether histone modifications can be inherited to the next generation. Furthermore, our data revealed a highly unique and non-canonical epigenome in early development that is distinct from those in somatic cells and embryonic stem cells.
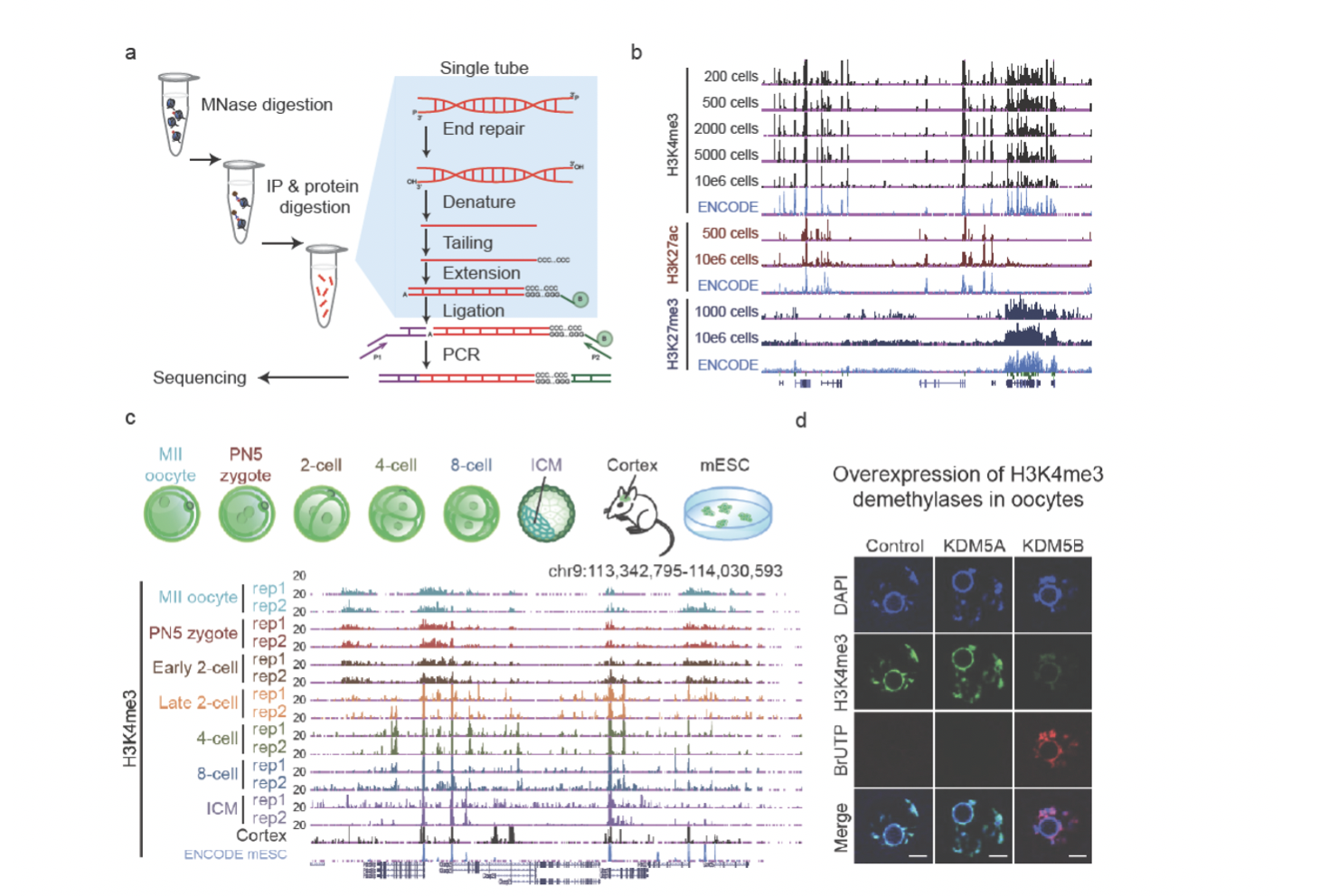
Figure 2. a, A brief schematic of STAR ChIP-seq. b, A snapshot of the UCSC browser view shows enrichment of STAR ChIP-seq for histone modifications using various numbers of mESCs. c, The UCSC browser view shows enrichment of H3K4me3 signals in early embryos (with replicates), the cortex, and mESCs (ENCODE). d, Immunostaining of H3K4me3 (green) and BrUTP (red, indicating transcription) in GV oocytes with overexpression of KDM5A or KDM5B by mRNA microinjection. Uninjected oocytes were used as a control. DAPI stains for DNA (blue). Figures are adapted from Zhang et al., Nature, 2016.
3. H3K4me3/H3K27me3 bivalent marks at promoters undergo an “erase-and-rewrite” process in early mouse development (Zheng et al., Molecular Cell, 2016).
In a separate study, we examined the inheritance and reprogramming in early development for another key histone mark H3K27me3, a classic repressive mark that plays crucial roles in regulating developmental genes. Interestingly, we again found H3K27me3 from the paternal genome is rapidly erased upon fertilization, while that from the maternal genome undergoes both inheritance and reprogramming. Specifically, H3K27me3 at promoters is erased from its targets such as the Hox genes in mouse preimplantation development. By contrast, H3K27me3 in distal regions appears to be inherited after fertilization and persists to blastocyst. Notably, it has been shown that developmental gene promoters are usually marked by both H3K4me3 and H3K27me3, known as “bivalent promoters” (Bernstein et al., 2006). Bivalent promoters are proposed to be poised for future activation. Interestingly, bivalent promoters are highly conserved among different species, and are also found in sperm (Hammoud et al., 2009; Lesch et al., 2016; Vastenhouw and Schier, 2012), raising an intriguing question whether bivalent marks can be inherited to early embryos to facilitate future activation of these key regulators of embryogenesis. However, our data showed that both H3K4me3 and H3K27me3 are erased from promoters of bivalent genes upon fertilization in mouse embryos. They are not readily established at these promoters until implantation. Thus, these data indicate an “erase-and-rewrite” mechanism for parental epigenetic memories carried by the bivalent marks during preimplantation development.
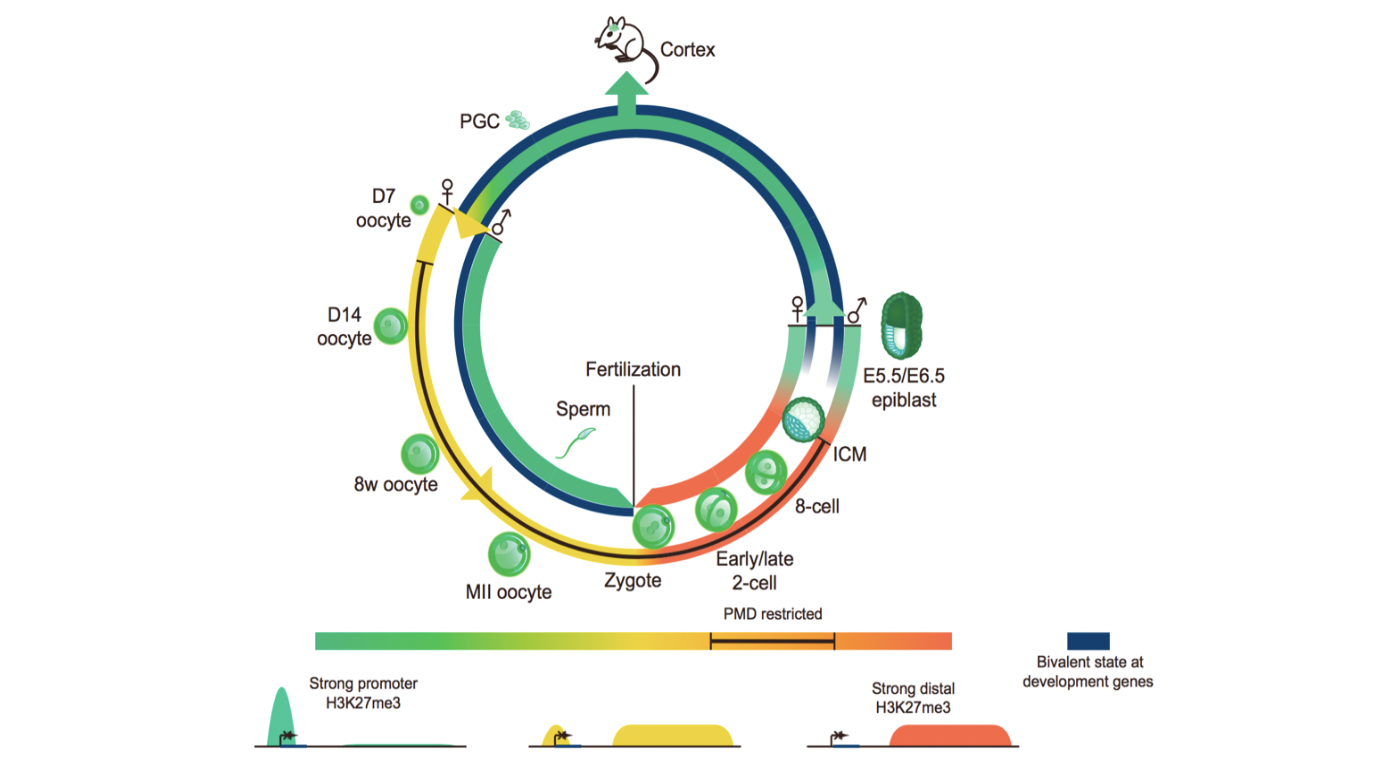
Figure 3. A schematic model showing dynamic regulation of H3K27me3 and bivalent marks in mouse gametogenesis and early development. A canonical form of H3K27me3, as well as the H3K4me3/H3K27me3 bivalent marks, exists in postimplantation embryos, PGCs (Sachs et al., 2013), and male germ cells (Hammoud et al., 2014) with strong enrichment at developmental gene promoters. A non-canonical form of H3K27me3 exists in oocytes and preimplantation embryos, with weak or no promoter H3K27me3 at developmental genes and relatively high enrichment of distal H3K27me3. H3K27me3 in oocytes after the onset of DNA methylome and the maternal alleles of preimplantation embryos is also shaped by oocyte PMDs. Figure is adapted from Zheng et al., Molecular Cell, 2016.
4. Genomic imprints in mice are regulated by a simple isoform switch of TET1 (Zhang et al., Molecular Cell, 2016).
Genomic imprints are DNA methylation marks that can be stable inherited from parents to progeny as epigenetic memories. Such parental memories are essential for embryogenesis and ensure that both parental genomes need to be present to allow successful development. Genomic imprints then will be erased in PGCs, to allow the re-establishment of parent-specific imprints in gametes for the next generation. In this work, we found two distinct isoforms for TET1, a methylcytosine oxidase, in mice. The full-length TET1 isoform (TET1e) is restricted to early embryos, embryonic stem cells (mESCs) and primordial germ cells (PGCs). By contrast, a short TET1 isoform (TET1s) is preferentially expressed in somatic cells, which lacks the N-terminus including the CXXC domain, a DNA-binding module. Unexpectedly, TET1s shows a similar binding pattern genomewide to that of TET1e, with preference to CpG islands (CGI) and promoters. Interestingly, further biochemistry assays showed that TET1e has significantly higher global chromatin affinity than that of TET1s. Furthermore, the global chromatin binding, but not targeted binding at CGIs, correlates with TET1-mediated demethylation. Finally, we carefully generated a mouse model with exclusive expression of Tet1s during the whole life cycle. Consequently, we found genomic imprints cannot be properly erased in PGCs of the mutant mice. The failure to do so in these mutant mice resulted in developmental defects of their progenies including decreased body weight and partial lethality. In summary, our data showed epigenetic memory between generations is regulated by a simple isoform switch of TET1 in mouse.
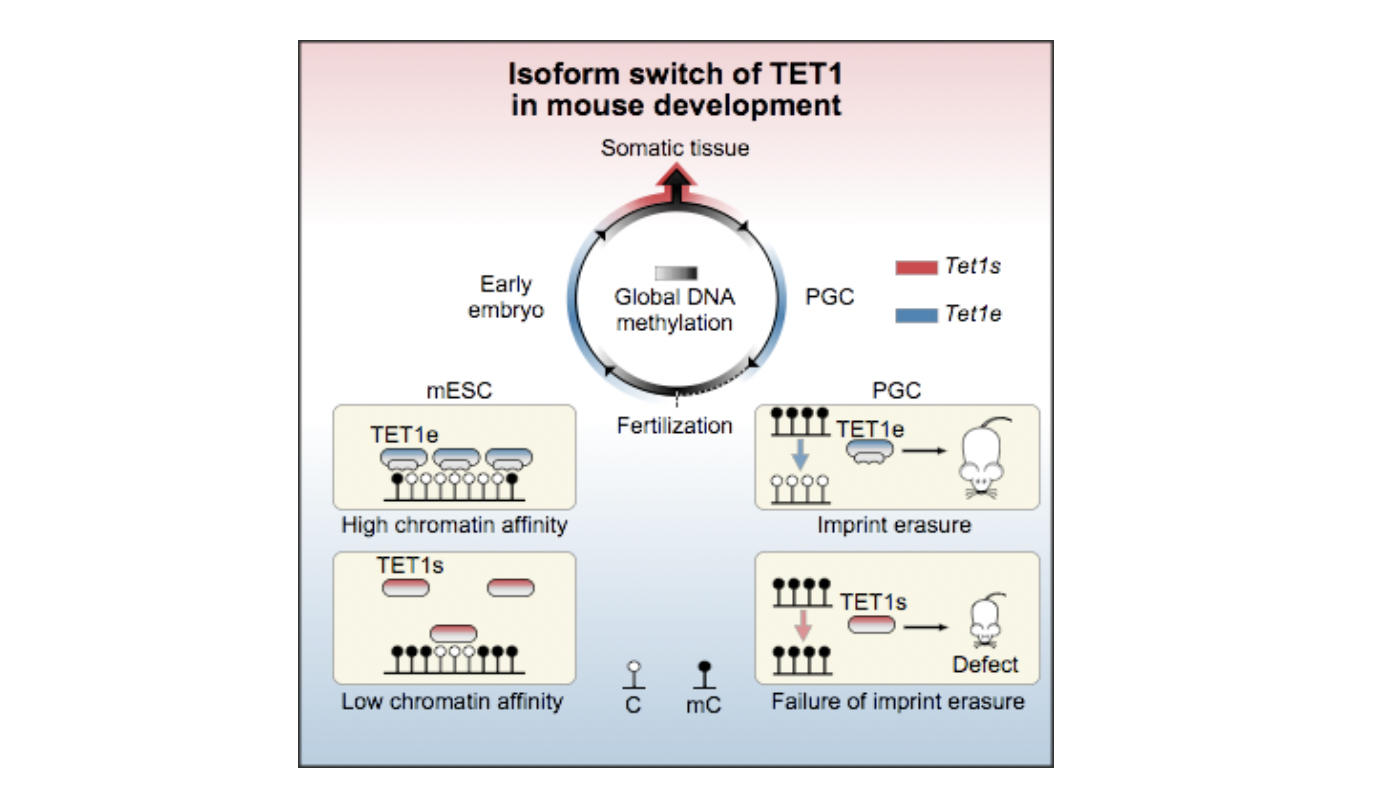
Figure 4. Isoform switch of TET 1 regulates DNA demethylation and genomic imprints. Tet 1 is expressed as a full-length isoform (Tet 1 e) in early embryos and PGCs, where global demethylation occurs. By contrast, Tet 1 is expressed as an isoform (Tet 1 s) that lacks the N terminus, including the CXXC domain, in somatic tissues (left). TET 1 e shows higher chromatin affinity compared with TET 1 s and leads to stronger DNA demethylation in mESCs. In PGCs, the exclusive expression of Tet 1 s instead of Tet 1 e results in defects in imprint erasure. Such a deficiency can be transmitted to the next generation, causing aberrant imprinted gene expression and developmental defects. Figure is adapted from Zhang et al., Molecullar Cell, 2016.
5. We showed how 3D chromatin structure is reprogrammed in early mammalian development (Du et al., Nature, 2017).
In eukaryotes, the genetic material DNA does not exist in a linear manner, but instead exists in the nucleus in a certain three-dimensional structure. The 3D chromatin structure plays an important role in biological processes such as gene expression regulation, DNA replication and cell division. However, the three-dimensional structure of chromatin and its reprogramming in preimplantation development remain poorly understood. Here, by developing a low-input Hi-C (genome-wide chromosome conformation capture) approach, we examined the reprogramming of chromatin organization during early development in mice. We found that oocytes in metaphase II show homogeneous chromatin folding that lacks detectable topologically associating domains (TADs) and chromatin compartments. Strikingly, chromatin showed greatly diminished higher-order structure after fertilization. Unexpectedly, the subsequent establishment of chromatin organization is a prolonged process that extends through preimplantation development, as characterized by slow consolidation of TADs and segregation of chromatin compartments. The two sets of parental chromosomes are spatially separated from each other and display distinct compartmentalization in zygotes. Such allele separation and allelic compartmentalization can be found as late as the 8-cell stage. Finally, we showed that chromatin compaction in preimplantation embryos can partially proceed in the absence of zygotic transcription and is a multi-level hierarchical process. Taken together, our data suggest that chromatin may exist in a markedly relaxed state after fertilization, followed by progressive maturation of higher-order chromatin architecture during early development.
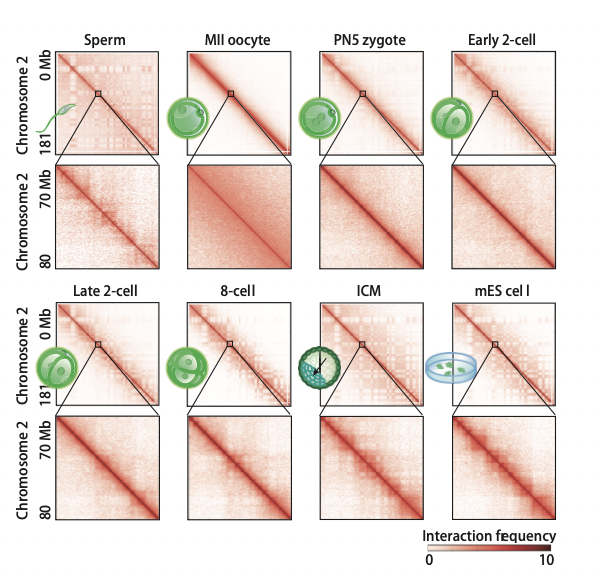
Figure 5. Heatmaps showing normalized Hi-C interaction frequencies (100-kb bin, chromosome 2) in mouse gametes and preimplantation embryos. Zoomed-in views (40-kb bin) are also shown. Figures are adapted from Du et al., Nature, 2017.
6. We revealed how chromatin is reprogrammed at the DNA levels in human early embryos (Wu et al., Nature, 2018).
Despite the progress in understanding chromatin reprogramming in mouse early development, epigenetic reprogramming in human embryos remains poorly understood. By developing an improved assay for transposase-accessible chromatin with high-throughput sequencing (mini-ATAC-seq), we investigate chromatin states in human preimplantation development. Integrative analyses show both conservation and divergence in regulatory circuitry between human and mouse early development, and between human pluripotency in vivo and human embryonic stem cells. In addition, we find widespread open chromatin regions before zygotic genome activation (ZGA). The accessible chromatin loci are readily found at CpG-rich promoters. Unexpectedly, many others reside in distal regions that overlap with DNA hypomethylated domains in human oocytes and are enriched for transcription factor-binding sites. A large portion of these regions then become inaccessible after ZGA in a transcription-dependent manner. Notably, such extensive chromatin reorganization during ZGA is conserved in mice and correlates with the reprogramming of the non-canonical histone mark H3K4me3, which is uniquely linked to genome silencing. Taken together, these data not only reveal a conserved principle that underlies the chromatin transition during mammalian ZGA, but also help to advance our understanding of epigenetic reprogramming during human early development and in vitro fertilization.
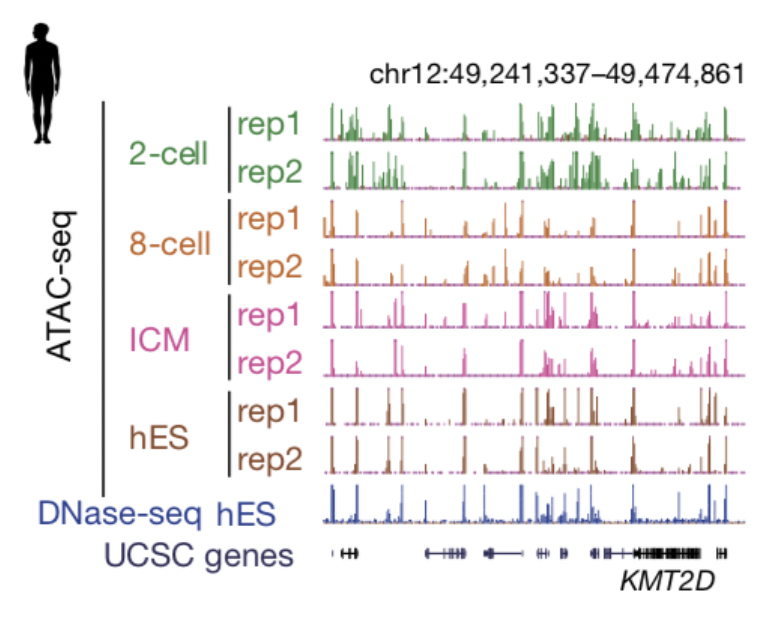
Figure 6. The UCSC browser view shows enrichment of ATAC-seq in human preimplantation embryos. Figures are adapted from Wu et al., Nature, 2018.
7. We demonstrated how the zygotic epigenome is established during early embryonic lineage determination in mammals (Zhang et al., Nature Genetics, 2018).
In mammals, early lineage specification in preimplantation and postimplantation embryonic development generates founder tissues for all subsequent somatic development. However, because of the limited materials available and the difficulty of tissue isolation in early embryos, lineage-specific regulation of transcriptomes and epigenomes in peri- and postimplantation embryos is poorly characterized. By developing a low-input method for genome-wide DNA-methylation profiling STEM-seq (small-scale TELP-enabled methylome sequencing), we examined the DNA methylation and chromatin structure in 11 lineage tissues in peri- and postimplantation mouse embryos (E3.5 to E7.5). We found allele-specific and lineage-specific de novo methylation at CG and CH sites that led to differential methylation between embryonic and extraembryonic lineages at promoters of lineage regulators, gene bodies, and DNA-methylation valleys. By using Hi-C experiments to define chromatin architecture across the same developmental period, we demonstrated that both global demethylation and remethylation in early development correlate with chromatin compartments. Dynamic local methylation was evident during gastrulation, which enabled the identification of putative regulatory elements. Finally, we found that de novo methylation patterning does not strictly require implantation. These data revealed dynamic transcriptomes, DNA methylomes, and 3D chromatin landscapes during the earliest stages of mammalian lineage specification.
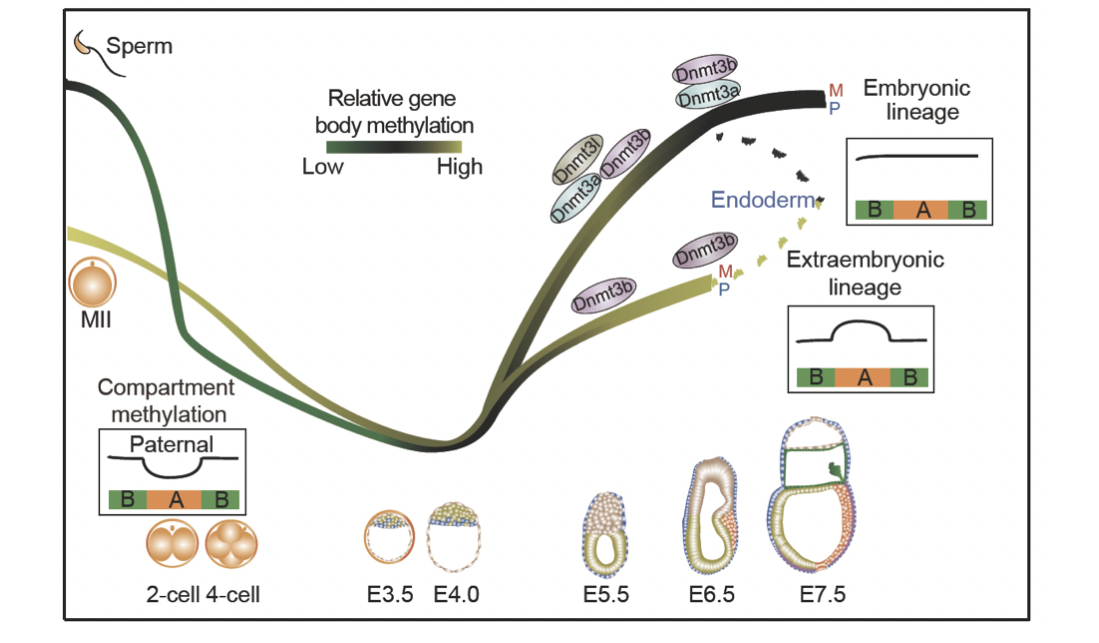
Figure 7. Dynamic regulation pattern of DNA methylation and chromatin advanced structure (display interval compartment A/B) in early mouse lineage differentiation. Figures are adapted from Zhang et al., Nature Genetics, 2018.